Descubren como ayudar a las células "cansadas" en enfermos mitocondriales




Cell - 19 de septiembre de 2013
- El hallazgo, que se publica en la revista Cell, demuestra que el gen OPA1 podría ser potencialmente útil para futuros tratamientos de muchas enfermedades mitocondriales, hasta ahora incurables
- Se trata un gen responsable de una enfermedad hereditaria, la atrofia óptica dominante, caracterizada por un funcionamiento incorrecto de la mitocondria, la encargada de suministrar la energía que necesitan las células para funcionar
Un equipo de investigadores del Centro Nacional de Investigaciones Cardiovasculares (CNIC) y de la Universidad de Padua publica en la última edición de la revista Cell un hallazgo que podría cambiar el futuro de los afectados por las enfermedades mitocondriales, un grupo de patologías caracterizadas por el mal funcionamiento de la mitocondria, la encargada de suministrar la energía que necesitan las células para funcionar.
La clave la ofrece el gen OPA1 que, cuando está alterado, es el responsable de una enfermedad hereditaria oftalmológica, la atrofia óptica dominante, que se caracteriza por una pérdida de agudeza visual insidiosa bilateral y simétrica en la primera o segunda década de la vida.
Los grupos del Dr. Luca Scorrano, profesor de Bioquímica en la Universidad de Padua y el Dr. José Antonio Enríquez, coordinador del Programa de Homeostasis y Reparación Celular del CNIC, han estudiado de cerca a este gen, deduciendo que tiene capacidad como un potencial ayudante del metabolismo celular, lo que podría ser explotado para desarrollar futuros tratamientos en el ámbito de las enfermedades mitocondriales, muchas de las cuáles no tienen cura.
“Las mitocondrias están en todas nuestras células y en ellas se encargan de regular aspectos tan importantes como la producción de la energía a partir de los alimentos o la preparación de la célula para dividirse, diferenciarse o incluso morir si es lo que resulta mas adecuado”, explica el Dr. Enriquez.
Durante años nuestros grupos han estudiado de forma independiente el funcionamiento de las mitocondrias y sus enfermedades, con intención de encontrar claves para desarrollar terapias para las enfermedades mitocondriales específicamente y para comprender como la mitocondria esta implicada en patologías de alta incidencia”, añade el Dr. Scorrano.
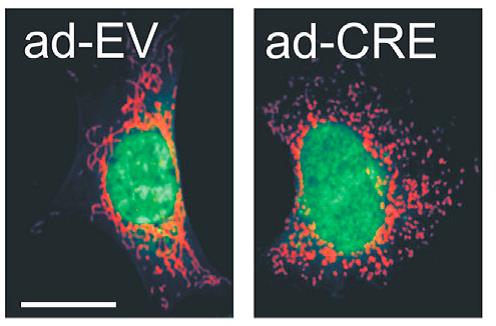
Hace cinco años decidimos aunar esfuerzos entre los dos grupos para tratar de entender una de ellas, la atrofia óptica dominante. Los afectados por la enfermedad oftalmológica presenta alteraciones en el gen OPA1, que contiene información de una proteína que ha sido caracterizada en los últimos años por el grupo italiano y que se encarga de regular la forma de la mitocondria. Su ausencia en los pacientes se traduce en la muerte progresiva de un tipo de neurona, las células ganglionares de la retina, responsables de la transmisión de imágenes desde el ojo a la porción del cerebro encargada de procesarlas. Esta pérdida de neuronas y, consecuentemente, de visión, es lenta pero progresiva: la enfermedad se suele manifestar en la edad prescolar, con distintos niveles de gravedad y siempre dentro de una misma familia. El grupo español había desarrollado nuevos modelos para el estudio de la función mitocondrial que parecían adecuados para entender la función del gen OPA1.
"Lo que hemos demostrado con este trabajo es que la tarea de OPA1 es regular la eficiencia de la respiración, influyendo en la manera en la que los componentes de lo que nosotros llamamos cadena respiratoria (por ejemplo, los complejos de proteínas que transforman los nutrientes en energía que pueden ser utilizados para actividades celulares) interactúan conjuntamente en la membrana interna de la mitocondria”, explican los doctores Enriquez y Scorrano.
Esta membrana es como un fluido, una línea escarpada que puede cambiar constantemente según los estímulos pero en la que, no obstante, los pliegues no se presentan por casualidad sino que están determinados por la actividad del gen OPA1.
Lo que los investigadores han demostrado es que incrementando la actividad de esta proteína se pueden mejorar la eficiencia de la cadena respiratoria a la hora de producir energía y de hacer crecer las células. “Somos capaces de pensar en un futuro en el que esta capacidad se explote como intervención terapéutica en distintas enfermedades mitocondriales, mejoran el metabolismo con independencia del defecto genético responsable de la disfunción mitocondrial”, añade el investigador.
“Para enfermedades heterogéneas y raras como estas, debemos descubrir aproximaciones terapéuticas más amplias, que se puedan aplicar a varias enfermedades a la vez. Estamos trabajando en este sentido, pero todavía es muy pronto para hablar de un posible tratamiento”, apunta el Dr. Scorrano.
“Nuestro trabajo supone un avance importante en el conocimiento que tenemos de la relación entre la función y la forma de las estructuras celulares. En particular, la estructura en cresta de la mitocondria es muy peculiar y sorprendentemente varialble entre tejidos, actividad, régimen de alimentación, patología. Sin embargo, la relación entre la forma variable de esta cresta y su actividad bioenergética no ha estado clara por muchos años. Este trabajo demuestra que cambiar dicha estructura influye en la capacidad bioenergética de la mitocondria a través de la modulación de súper complejos respiratorios”, indicar por su parte el Dr. Enríquez.
Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency
Cell, 2013; DOI: 10.1016/j.cell.2013.08.032
Sara Cogliati, Christian Frezza, Maria Eugenia Soriano, Tatiana Varanita, Ruben Quintana-Cabrera, Mauro Corrado, Sara Cipolat, Veronica Costa, Alberto Casarin, Ligia C. Gomes, Ester Perales-Clemente, Leonardo Salviati, Patricio Fernandez-Silva, Jose A. Enriquez, Luca Scorrano.



